[1]国家统计局. 国家统计局关于2024年粮食产量数据的公告[EB/OL].(2024‑12‑13)[2025‑11‑18].https://www.stats.gov.cn/sj/zxfb/202412/t20241213_1957744.html.
National Bureau of Statistics of China.Announcement of National Bureau of Statistics of China on the grain output data of 2024[EB/OL]. (2024‑12‑13)[2025‑11‑18].https://www.stats.gov.cn/sj/zxfb/202412/t20241213_1957744. html.
[2]王晓鸣,段灿星. 玉米病害和病原名称整理及其汉译名称规范化探讨[J]. 中国农业科学,2020,53(2):288‑316.
WANG X M,DUAN C X. Reorganization of maize disease and causal agent names and discussion on their standardized translation of Chinese names[J]. Scientia Agricultura Sinica,2020,53(2):288‑316.
[3]贺字典,陈捷,高增贵,等. 玉米丝黑穗病及病菌生理分化研究进展[J]. 玉米科学,2005,13(4):117‑120.
HE Z D,CHEN J,GAO Z G,et al. Progress on studies of head smut physiological differential in maize[J].Journal of Maize Sciences,2005,13(4):117‑120.
[4]王振华,姜艳喜,王立丰,等. 玉米丝黑穗病的研究进展[J]. 玉米科学,2002,10(4):61‑64.
WANG Z H,JIANG Y X,WANG L F,et al. Research advance on head smut disease in maize[J].Maize Sciences,2002,10(4):61‑64.
[5]晋齐鸣,王晓鸣,王作英,等.东北春玉米区玉米丝黑穗病大发生原因及对策[J]. 玉米科学,2003,11(1):86‑87.
JIN Q M,WANG X M,WANG Z Y,et al. The epidemiological factors and control tactics of head smut in spring corn area of northeast of China[J]. Journal of Maize Sciences,2003,11(1):86‑87.
[6]王晓鸣. 玉米病虫害田间手册[M]. 北京:中国农业科技出版社,2002.
WANG X M. Field manual of maize diseases and insect pests[M]. Beijing:China Agricultural Science and Technology Press,2002.
[7]王富荣,石秀清,石银鹿. 山西省玉米病害的发生现状及防治对策[J]. 玉米科学,2000,8(3):79‑80.
WANG F R,SHI X Q,SHI Y L. Occurrence and control of maize disease in Shanxi province[J]. Journal of Maize Sciences,2000,8(3):79‑80.
[8]WANG Y,XU J,DENG D,et al. Transcriptomic and weighted gene co‑expression network analysis reveals candidate genes for bacterial leaf streak resistance in maize[J]. The Plant Journal,2020,102(4):748‑763.
[9]马亮,马璐,张舒钰,等. 玉米苞叶数目转录组分析及候选基因鉴定[J/OL]. 作物学报,2025[2025‑12‑21].https://link.cnki.net/urlid/11.1809.S.20251121.1008.002.
MA L,MA L,ZHANG S Y,et al. Transcriptome analysis of husk leaf number in maize and identification of candidate genes[J/OL]. Acta Agronomica Sinica,2025[2025‑12‑21].https://link.cnki.net/urlid/11.1809.S.20251121.1008.002.
[10]周超,王俊强,韩业辉,等. 玉米抗穗腐病候选基因的克隆及分子标记开发[J]. 江苏农业科学,2025,53(19):38‑44.
ZHOU C,WANG J Q,HAN Y H,et al. Cloning of candidate genes for maize ear rot resistance and development of molecular markers[J]. Jiangsu Agricultural Sciences,2025,53(19):38‑44.
[11]LANGFELDER P,HORVATH S. WGCNA:An R package for weighted correlation network analysis[J].BMC Bioinformatics,2008,9(1):559.
[12]ZHANG X,LI Z,ZHANG H,et al. Weighted gene co‑expression network analysis identifies hub genes associated with resistance to head smut in maize[J].Molecular Plant Pathology,2022,23(1):123‑135.
[13]刘健茁. 玉米-瘤黑粉菌互作中光合相关基因挖掘[D]. 沈阳:沈阳农业大学,2025.
LIU J Z. Mining photosynthesis‑related genes in corn‑smut interaction[D]. Shenyang:Shenyang Agricultural University,2025.
[14]高刚强. 基于转录组和代谢组解析玉米抗小斑病分子机理[D]. 郑州:河南农业大学,2024.
GAO G Q. Molecular mechanism of maize resistance to southern leaf blight based on transcriptome and metabolome analysis[D].Zhengzhou:Henan Agricultural University,2024.
[15]申涛.玉米自交系T32抗丝黑穗病主效QTL的精细定位[D]. 贵阳:贵州大学,2022.
SHEN T. Fine mapping of major QTLs for resistance to head smut in maize inbred line T32[D]. Guiyang:Guizhou University,2022.
[16]周天旺,王春明,洪流,等. 331份玉米自交系对丝黑穗病的抗性鉴定与评价[J].玉米科学,2024,32(3):101‑106.
ZHOU T W,WANG C M,HONG L,et al.Identification and evaluation of 331 maize germplasm resistance to Sporisorium reilianum causing head smut [J]. Journal of Maize Sciences,2024,32(3):101‑106.
[17]郭致杰,李国权,周天旺,等. 58份鲜食玉米品种对瘤黑粉病和丝黑穗病的抗性评价[J]. 玉米科学,2023,31(6):143‑150.
GUO Z J,LI G Q,ZHOU T W,et al. Evaluation of resistance to Ustilago maydis and Sporisorium reilianum in 58 fresh maize varieties[J].Journal of Maize Sciences,2023,31(6):143‑150.
[18]郭成,张小杰,王春明,等. 42份鲜食玉米品种对丝黑穗病和瘤黑粉病的抗性[J]. 西北农业学报,2021,30(9):1427‑1433.
GUO C,ZHANG X J,WANG C M,et al. Resistance of 42 fresh‑eating maize varieties to Sporisorium reilianum and Ustilago maydis[J]. Acta Agriculturae Boreali‑Occidentalis Sinica,2021,30(9):1427‑1433.
[19]刘玲玲,韩苗苗,王乾坤,等. 基于吉846/掖3189重组近交系群体的玉米丝黑穗抗性的QTL分析[J]. 玉米科学,2016,24(3):20‑25.
LIU L L,HAN M M,WANG Q K,et al.Density‑increasing of genetic map and QTL analysis for maize head smut resistance based on Ji846/Ye3189 RIL population[J]. Journal of Maize Sciences,2016,24 (3):20‑25.
[20]QIU H B,TAN K,LI C H,et al. Identification of QTL for resistance to head smut in maize(Zea mays. L)[J].Euphytica,2021,217(9):185.
[21]ZHOU Y,YAO M H,WANG Q,et al. Analysis of QTLs and candidate genes for tassel symptoms in maize infected with Sporisorium reilianum[J].International Journal of Molecular Sciences,2022,23 (22):14416.
[22]ZHANG L,ZHOU X,SUN X H,et al. Development of a dCAPS marker based on a minor resistance gene related to head smut in BIN8. 03 in maize[J].Pakistan Journal of Botany,2020,52(5):1849‑1855.
[23]DI H,DENG Y X,GUO X H,et al. Prediction of a candidate resistance gene and development of the LSdCAP8 marker related to head smut resistance in maize[J]. Pakistan Journal of Botany,2018,50(1):399‑406.
[24]刘潮,韩利红,王海波,等. 植物类甜蛋白基因家族研究进展[J]. 生物技术通报,2018,34(3):9‑17.
LIU C,HAN L H,WANG H B,et al. Research advances on plant thaumatin‑like protein family[J].Biotechnology Bulletin,2018,34(3):9‑17.
[25]张绍鹏. 玉米抗丝黑穗病的分子机理研究[D]. 武汉:华中农业大学,2012.
ZHANG S P. Studies on the molecular mechanism of head smut resistance in maize(Zea mays L.)[D].Wuhan:Huazhong Agricultural University,2012.
[26]高树仁. 玉米抗丝黑穗病遗传分析及数量性状基因定位[D]. 长春:吉林大学,2005.
GAO S R. Inheritance and quantitative trait loci mapping of resistance to head smut caused by Sphacelotheca reiliana(Kühn) in maize[D].Changchun:Jilin University,2005.
[27]章志强. 植物WAK家族起源和进化及GhWAKL39调控棉花衣分的分子机制[D]. 武汉:华中农业大学,2023.
ZHANG Z Q. Origin and evolution of plant WAK family and molecular mechanism of GhWAKL39 regulating cotton lint percentage[D]. Wuhan:Huazhong Agricultural University,2023.
[28]左为亮. 玉米抗丝黑穗病主效QTL的克隆与抗病机理研究[D]. 北京:中国农业大学,2014.
ZUO W L. Cloning and characterization of a major QTL conferring[D]. Beijing:China Agricultural University,2014.
[29]ZHANG B Q,ZHANG N,ZHANG Q Q,et al.Transcriptome profiles of Sporisorium reilianum during the early infection of resistant and susceptible maize isogenic lines[J]. Journal of Fungi,2021,7(2):150.
[30]ZHANG Q Q,XU Q Y,ZHANG N,et al. A maize WAK‑SnRK1α2‑WRKY module regulates nutrient availability to defend against head smut disease[J].Molecular Plant,2024,17(11):1654‑1671.
[31]王明. 玉米抗丝黑穗病的全基因组关联分析[D]. 武汉:华中农业大学,2012.
WANG M. Genome‑wide association study(GWAS)of resistance to head smut in maize(Zea mays L.)[D].Wuhan:Huazhong Agricultural University,2012.
[32]GHAREEB H,BECKER A,IVEN T,et al. Sporisorium reilianum infection changes inflorescence and branching architectures of maize[J]. Plant Physiology,2011,156(4):2037‑2052.
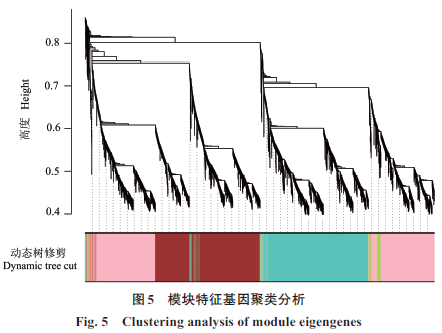
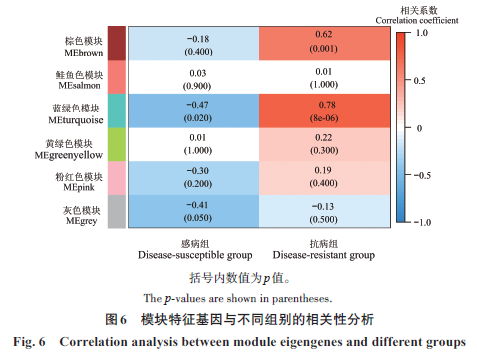
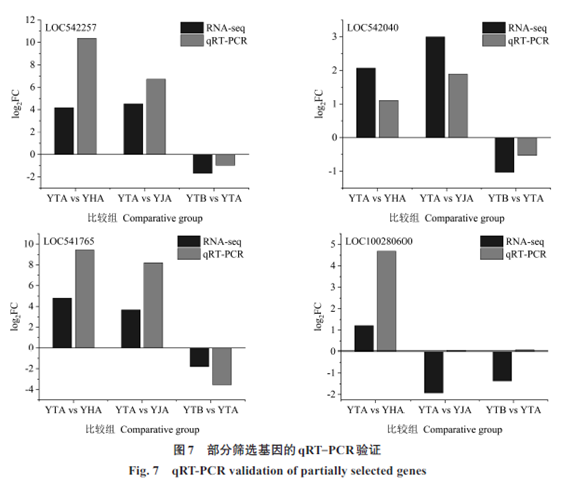
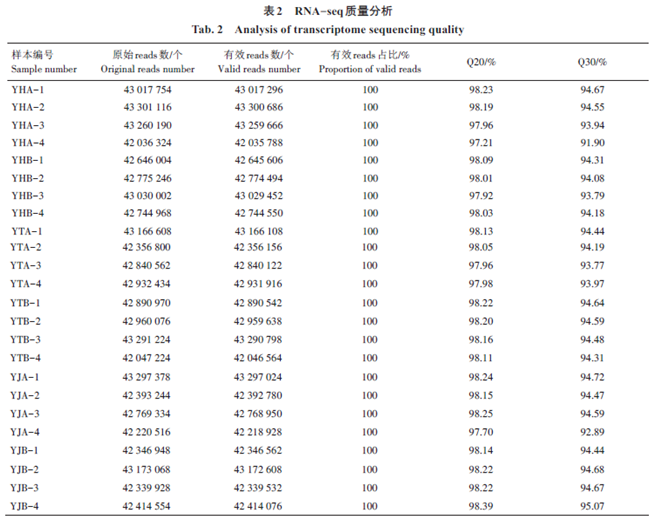
|